
Cause
Cystic Fibrosis is an inherited condition caused by a gene mutation. Even if parents don't show symptoms of CF, they can still be carriers and pass on the disease to their children.
Inheritance
Cystic Fibrosis (CF) is recessively inherited, which means that two copies of the mutated gene are required before a person will show symptoms of the disease. Therefore, for a baby to born with cystic fibrosis, both parents must be carriers. The diagram below shows the inheritance pattern of CF:
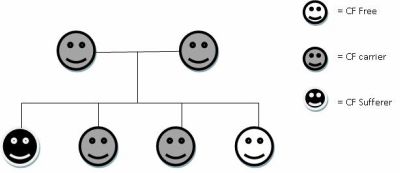
Each child born to two carriers has a 1 in 4 chance of developing the disease, a 2 in 4 chance of being a carrier and a 1in 4 chance of having no CF genes. The mutation in CF is loss of function, and lies in a gene coding for the cystic fibrosis transmembrane conductance regulator (CFTR) channel.
CFTR
A mutation in the gene encoding the CFTR chloride ion channel is one of the main causes of cystic fibrosis.
CFTR belongs to ATP-binding cassette transporter family. It makes a protein that functions as a membrane channel for chloride ions, and also regulates the function of other ion channels, such as sodium channels. Chloride ions passing through the CFTR channel also helps to control movement of water in tissues, which maintains the fluidity of secretions such as mucus.
CFTR is expressed in the lungs, pancreas, liver, secretory cells, sinuses and the reproductive tract. It also regulates other proteins and ion channels. When CFTR is synthesised, it is transported to the endoplasmic reticulum and golgi apparatus for additional processing before being integrated into the cell membrane.The diagram below shows a mutated CFTR ion channel causing cystic fibrosis.
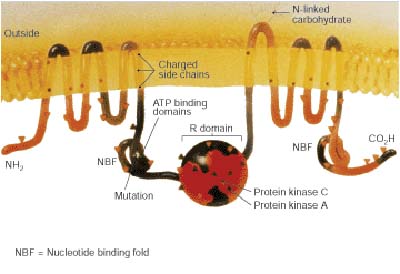
CFTR Mutation
 Mutations in CFTR which cause cystic fibrosis are in a gene located on chromosome 7 in locus q (see pic). A mutation in CFTR means that chloride ions can’t move across the epithelial membrane, and also has affects on other ions. Approximately 70% of mutations in CF patients result from deletion of three base pairs in CFTR's nucleotide sequence. This mutation is called delta F508.
Mutations in CFTR which cause cystic fibrosis are in a gene located on chromosome 7 in locus q (see pic). A mutation in CFTR means that chloride ions can’t move across the epithelial membrane, and also has affects on other ions. Approximately 70% of mutations in CF patients result from deletion of three base pairs in CFTR's nucleotide sequence. This mutation is called delta F508.
If a CFTR F508 mutation occurs, when it reaches the endoplasmic reticulum a quality control mechanism is activated which recognises the protein as being misfolded and marks the protein for degradation. This therefore leads to a reduction in the number of CFTR channels present on the cell membrane.
People who are homozygous for the F508 mutation tend to have the most severe symptoms of cystic fibrosis due to critical loss of chloride ion transport. This upsets the balance between sodium and chloride ions required to maintain the normal, thin mucus layer that is easily removed by the cilia lining the lungs and digestive tract. This imbalance causes the build up of thick, sticky mucus leading to cystic fibrosis symptoms.
Although the majority of patients with cystic fibrosis have a F508 mutation, there are many other mutations which can also cause the disease. There have been over 1,000 genes identified to date which, if mutated, lead to CF.
Images of CFTR channel https://commons.wikimedia.org/wiki/File:CFTR.jpg and CFTR chromosome 7 https://commons.wikimedia.org/wiki/Special:Search?search=CFTR courtesy of wikimedia commons under the GNU free documentation licence
{kind=link}